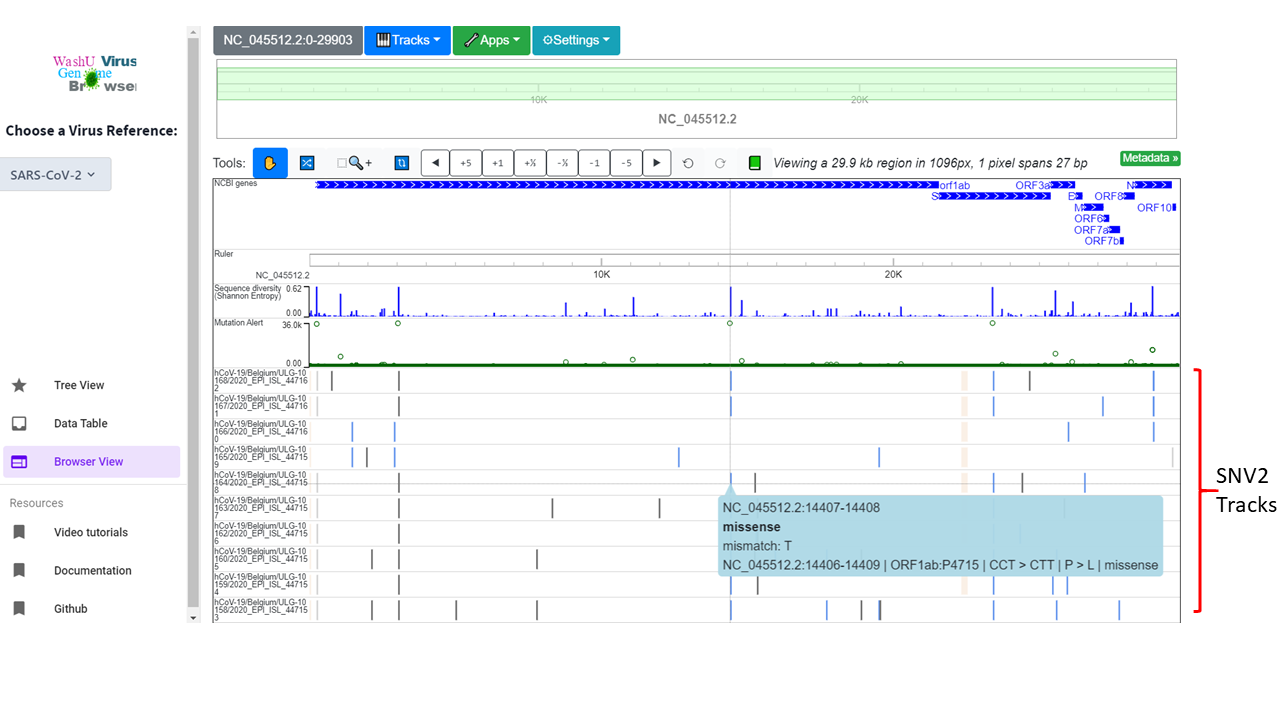
SNV2 track¶
Functionality of SNV2 tracks¶
SNV2 tracks are an extension of SNV tracks. It’s equipped with the ability to show amino acid level mutations. It also supports everything that SNV tracks support: color code mutations, zoomed-in and zoomed-out views, revealing detailed info upon mouse click, etc. SNV2 format is specified with extreme flexibility.
Essentially, the SNV2 format is a combination of categorical tracks (color coding) and bed tracks (text display). You can imagine it as “color coded bed tracks”. This enables it to be reused for many other purposes. We provide scripts to generate SNV2 tracks reporting the potential amino acid mutations, but we encourage you to customize it with your own scripts.
Defining the format of SNV2¶
SNV2 tracks can have as many columns as necessary. The first 3 columns encode the genomic positions. The 4th column encodes category. All columns starting from the 4th column will be shown as text upon mouse click. The mapping between categories and colors is specified in Json.
| column | detail |
|---|---|
| 1 | chromosome name for epigenome browser, reference name for virus genome browser (such as NC_045512.2 for SARS-CoV-2) |
| 2 | start position on the reference genome, 0 based, inclusive |
| 3 | stop position on the reference genome, 0 based, not inclusive |
| 4 | category. Controls color code, will also show as text in popup window upon clicking |
| 5, 6, 7… | text columns. will show as text in popup window upon clicking |
The 4th column is mapped to colors through specifying “segmentColors” in the “options” part of the json of datahubs. The detailed tutorial on how to use json is here: https://epigenomegateway.readthedocs.io/en/latest/datahub.html?highlight=json
However, if you are using the SNV2 track to show AA mutations, you don’t need to upload as json, because we have default color code mapping:
"options": {
"segmentColors": {
"un_sequenced": "Linen",
"noncoding_insertion": "LightGrey",
"noncoding_deletion": "LightGrey",
"noncoding_mismatch": "LightGrey",
"silent": "DimGrey",
"frameshift": "FireBrick",
"missense": "CornflowerBlue",
"AA_deletion": "CornflowerBlue",
"AA_insertion": "CornflowerBlue",
"N_mask": "Linen",
"deletion_mask": "Linen"
}
}
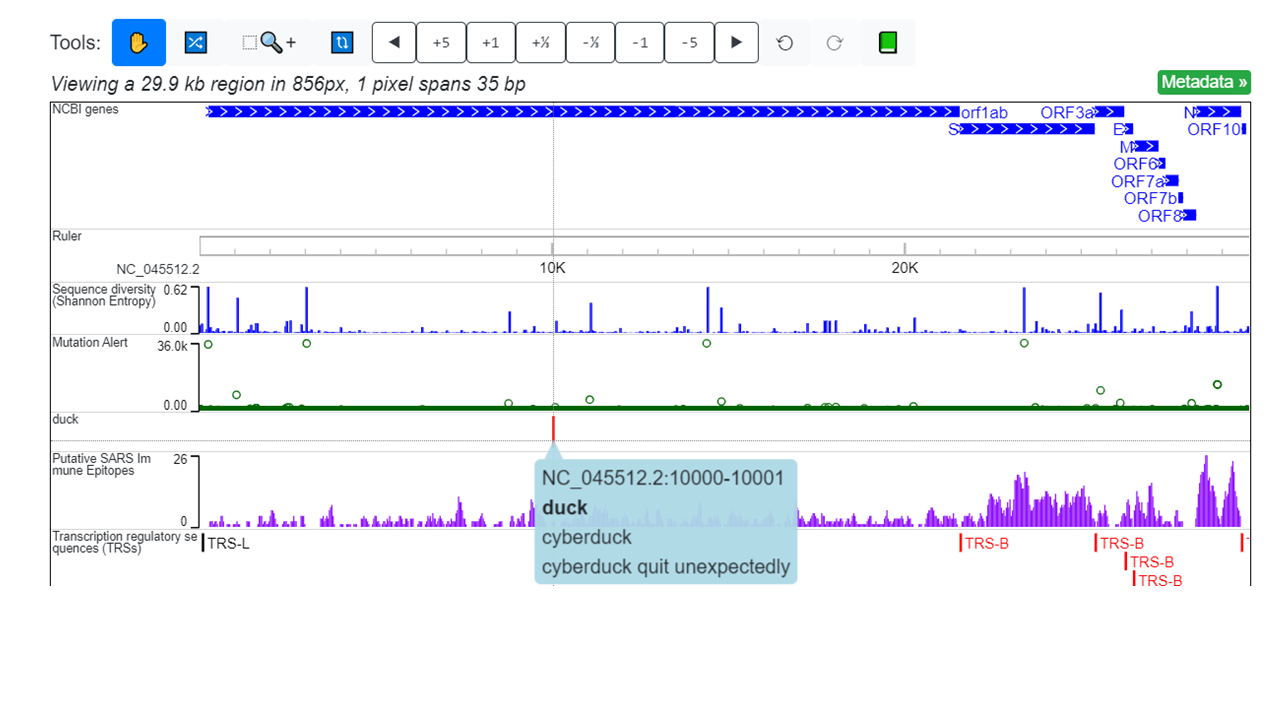
For a quick demo:
NC_045512.2 10000 10001 duck cyberduck cyberduck quit unexpectedly
zip it and index it using bgzip and tabix (https://epigenomegateway.readthedocs.io/en/latest/tracks.html?highlight=tabix#prepare-track-files). Then put it into a Json like this:
[{
"name": "duck",
"type": "snv2",
"url": "http://your.url.to.duck.file/duck.snv2.gz",
"options": {
"segmentColors": {
"duck": "red"
}
}
}]
upload the track through Tracks -> Remote Tracks -> Add Remote Data Hub You will see:
One of our snv2 tracks for SARS-CoV-2 is coded like this:
NC_045512.2 0 16 un_sequenced un_sequenced
NC_045512.2 240 241 noncoding_mismatch mismatch: T NC_045512.2:240-241 | ORF:noncoding | C > T | noncoding_mismatch
NC_045512.2 3036 3037 silent mismatch: T NC_045512.2:3034-3037 | ORF1ab:F924 | TTC > TTT | F > F | silent ; NC_045512.2:3034-3037 | ORF1a:F924 | TTC > TTT | F > F | silent
NC_045512.2 14407 14408 missense mismatch: T NC_045512.2:14406-14409 | ORF1ab:P4715 | CCT > CTT | P > L | missense
NC_045512.2 23402 23403 missense mismatch: G NC_045512.2:23401-23404 | S:D614 | GAT > GGT | D > G | missense
NC_045512.2 29872 29903 un_sequenced un_sequenced
Scripts for generating snv2 tracks¶
All of our premade SNV2 tracks that you can see in Tracks -> Public Data Hubs are generated through a set of scripts that can be found at https://github.com/debugpoint136/WashU-Virus-Genome-Browser/tree/master/scripts/snv2/
The main function tsv2snv2.2(), which we used to generate all snv2 files in the public data hubs, is in snv2_public_7_2_20.R, while the helper functions are in snv2_helper_7_2_20.R. We used snv2_orf_7_2_20.R to generate the tsv file with ORF information required in the main function.
The arguments of tsv2snv2.2():
| argument | detail |
|---|---|
| tsv.vec | a vector of tsv files generated by publicAlignment.py in tempt_dir (an argument for publicAlign.py). |
| ref.fa | name of the fasta file for the reference. The one for SARS-CoV-2 is here |
| ref.orf.table | a dataframe or the name of a tsv file containing this dataframe. the ORF information for the reference. the one for SARS-CoV-2 is already generated. Refer to it for formatting. |
| min.contig.head.tail | integer. used to mask the beginning and the end of the sequenced region, so that unsequenced regions won’t be treated as deletions. default is 15, which means the first 15 continuous non-deletions marks the beginning of the sequenced region. The same applies for the end of the sequenced region |
| out.snv2.vec | a vector of output snv2 file name |
| hier.df | a dataframe containing the ‘hierarchy’ of different types of mutations, can also be the name of a tsv file containing this dataframe. Sometimes one nucleotide can be used by more than one ORF. The mutation at this nucleotide might cause different types of amimo acid mutations for different ORFs. For example, a mutation can be silent for ORF A, but missense for ORF B. In this case, ‘missense’ will override ‘silent’ because of the settings in hier.df. Refer to hier.df.6.18.tsv for formatting |
| thread.master | the number of snv2 files to generate in parallel |
| thread.sub | the number of orfs to process in parallel |
| bedtools.path | deprecated. Just pass it something |
| return.df | bool. if the entire snv2 track should be returned as a dataframe. |